Software
Here you can find a few software packages that I've developed. Since I'm a big supporter of the open source initiative all the packages are available under permissive open source licenses. If you want to have a full overview of my coding activities check out my repositories:
mendeleev


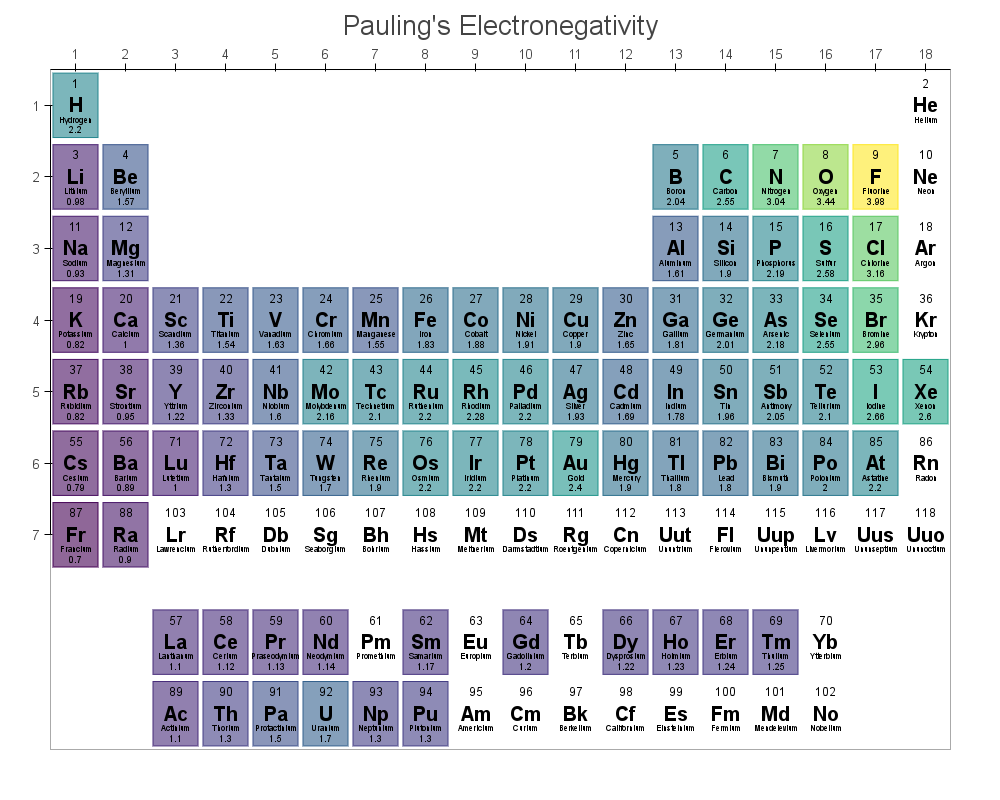
mendeleev is pythonic periodic table with a rich database of atomic, ionic and isotopic properties which is easy to use.
convenient methods to formulate complex querries, tranfer data to numpy and pandas objects and visualize various properties is color mapped periodic tables using variety of plotting backends like: bokeh, plotly or matplotlib.
All the atomic properties are listed in the data page of the documentation with their respective references. A particularly interesting feature is that the package includes 10 different electronegativity scales that are available directly or can be generated using appropriate functions from other stored properties.
panthera


A Package for ANharmonic THERmochemistry - a python implementation for calculating anharmonic corrections to thermochemical functions in the independent mode approximation for molecules and solids. The package was developed in collaboration with the group of Prof J. Sauer at Humboldt University in Berlin during my stay there and evolved from the work of G. Piccini. It is interfaced with the Atomistic Simulation Environment and should work with most of the programs supported by it.
Batch Calculator

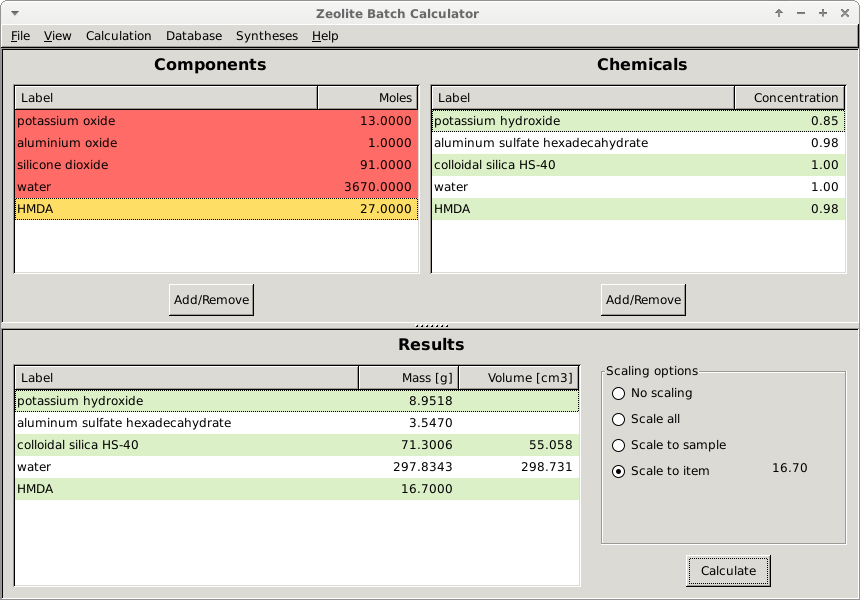
A desktop GUI app based on wxPython for calculating the correct amount of reactants (batch) for a particular composition given by the molar ratio of its components. It is designed to assist chemical synthesis when it requires batch preparation. Besides offering a quick and easy way of calculating the desired masses (or moles) it can also store information about chemicals, components and syntheses in a local database and generate lab reports in pdf format.
chemtools


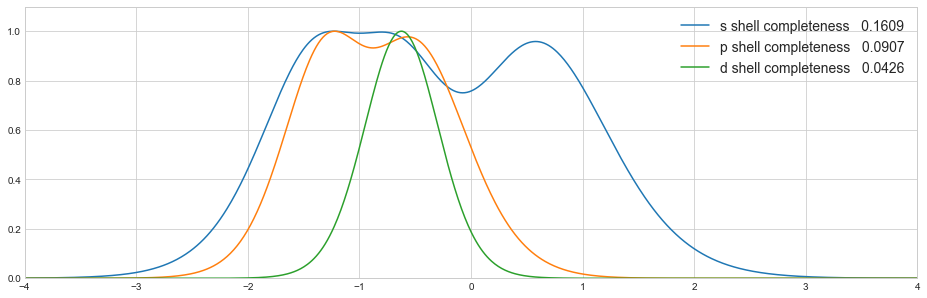
chemtools package evolved from a set of modules and scripts intended to help with automation of various tasks common in electronic structure calculations.
It provides a quite powerful BasisSet abstraction that allows one-particle basis sets to be handled efficiently. Some of the features include:- conversion between various basis set formats - input: GamessUS, Molpro and Gaussian - output: GamessUS, Molpro, Gaussian, NWChem, CFour/AcesII, Dalton
- optimization of primitive exponents using a wrapper around scipy.optimize module
- plotting basis set completeness profiles
- writing the input files,
- parsing the standard log files,
- parsing various parts of the PUNCH or .dat file
- reading the (direct access binary) dictionary file .F10 records into numpy arrays
- reading the (sequential unformatted binary) files with two-electron integrals (.F08, .F09), elements of the reduced two-particle density matrix (.F15, .F16), CI coefficients (.F12) into numpy arrays
ase-espresso


ase-espresso provides a Python interface compatible with Atomic Simulation Environment (ASE) for manging calculations with Quantum Espresso - the widely used open source package for plane wave Density Functional Theory (DFT) and molecular dynamics calculations.
The current version started from a fork from vossjo/ase-espresso and since then considerably diverged offering various improvements over the original version, the most important ones include:
- python 3.x compatible
- installable through pip
- documentation available through sphinx with a lot of docstrings updated
- the
site.cfgwas replaced by a newSiteConfigclass that dynamically gathers information about the execution environment - the old
espressoclass is now split into two:Espressopreserving the standard functionality andiEspressoresponsible for dynamic/interactive jobs with a custom version ofpw.x - the
Espressoclass were restructured according to ase guidelines regarding calculator objects to support full compatibility with ase - most of the system call are now handled by pexpect and subprocess instead of the old
os.system,os.popen(),os.popen2(),os.popen3() - tests were added
- code style and readability were improved
zefram

Is a convenience package for accessing ana analyzing various properties of zeolite frameworks combining the data available from:
in a python package allowing easy access to the stored data.colorcif

A small utility python script based on Atomistic Simulation Environment to generate high-quality images from Crystallographic Information File (CIF) files with symmetry unique atoms colore-mapped with different colors.